Navigate vast chemical space with generative de novo design
Quickly generate thousands of novel, synthesizable compounds while applying scaffold- and reaction-based constraints for more targeted exploration—expanding possibilities for breakthrough discovery.
Predict and prioritize hits with multi-parametric profiles
Refine and rank hit candidates with robust ADME, toxicity, and CYP metabolism predictions—powered by AI/ML models trained on 30+ years of preclinical data to help reduce DMTA cycles and increase success rates.
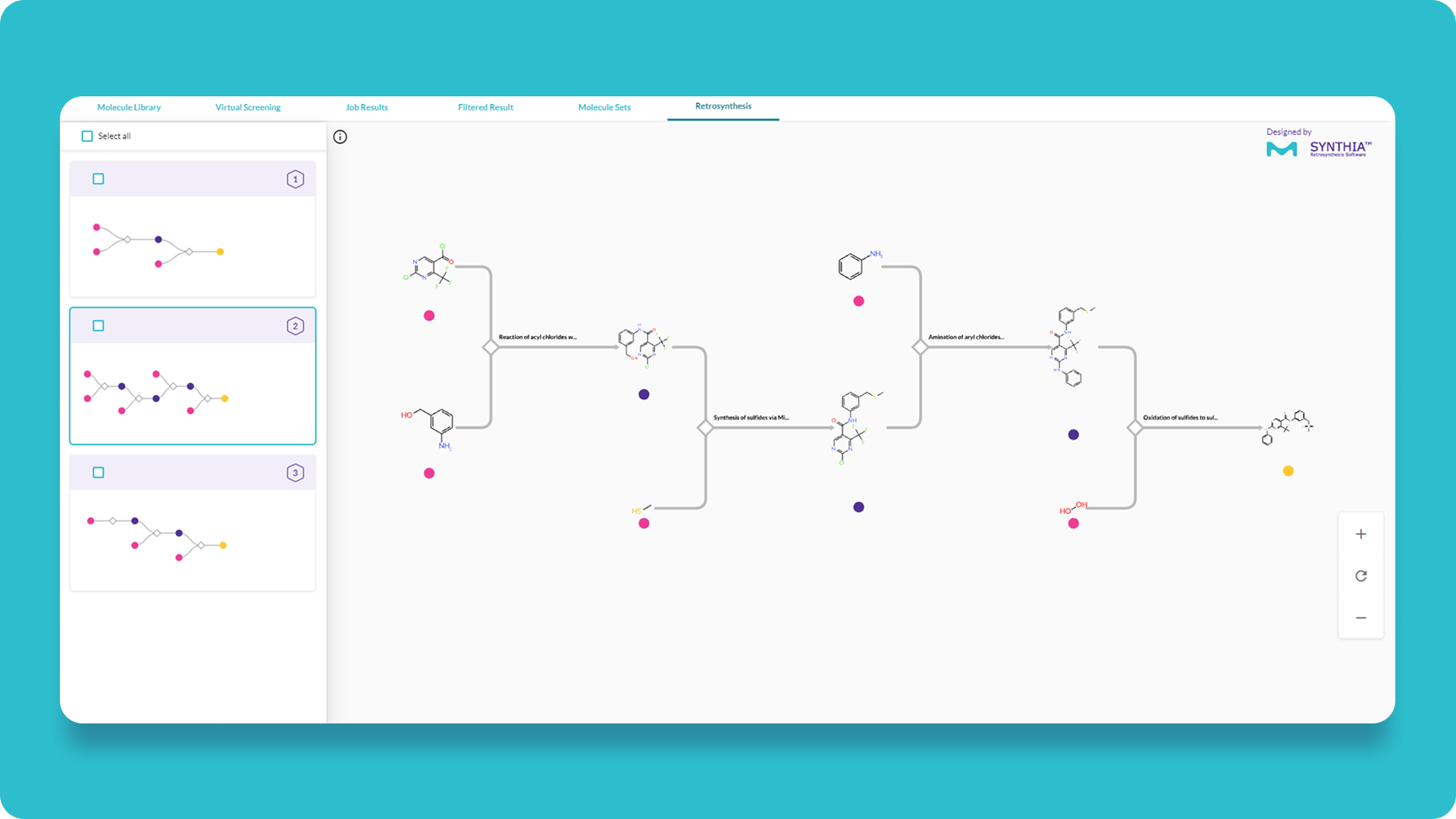
Streamline synthesis and chemical sourcing
Ensure synthetic feasibility from the start with built-in Synthetic Accessibility (SA) scores and retrosynthesis pathways, powered by SYNTHIA™’s 120,000+ reaction rules and integrated ordering via Aldrich® Market Select.
Seamless deployment and workflow integration
Deploy instantly with our cloud-based SaaS platform, eliminating IT complexity. Easily import and export molecules with CSV, SDF, and RD file formats, ensuring interoperability with your existing discovery tools.

Democratizing De Novo Molecule Design for Medicinal Chemists
No Coding Required
Designed for medicinal chemists—leverage generative AI without command-line prompts.
Intuitive & Transparent AI
Confidence indicators, glowing twins, and explainable AI ensure trustworthy insights.
Robust ADMET Endpoints
Trained on both positive and negative experimental data for reliable ADMET predictions.
Secure & Scalable
Keep your research and IP safe with ISO-27001 certified security and compliance standards.
GET STARTED
Find the Right Molecule with AIDDISON™
Ready to see how AIDDISON™ can transform your drug discovery? Contact us today to learn more and request a demo of our solutions.
Frequently Asked Questions
Resources
Explore our latest white papers, webinars, and more to stay informed about cutting-edge advancements in AI-powered drug discovery and retrosynthesis.


Videos
Discover how to apply generative AI methods in hit identification, hit-to-lead, and lead optimization of novel molecules.

White Papers
Learn how to unlock new chemical space with generative AI methods that expand options for drug discovery.

